Professor Jie Chen's group recently published a paper entitled "Accurate description of high-order phonon anharmonicity and lattice thermal conductivity from molecular dynamics simulations with machine learning potential".
Phonon anharmonicity is critical for accurately predicting the material’s thermal conductivity (κ). However, its calculation based on the perturbation theory is a difficult and time-consuming task, especially for the high-order phonon scattering process. In this work, using cubic boron arsenide (BAs) and diamond as examples, we combine the machine learning potential (MLP) with molecular dynamics simulations to predict κ and assess the effect of anharmonicity on thermal transport properties. A MLP based on the matrix tensor algorithm is developed in this work, which can accurately describe lattice dynamics behaviors in both BAs and diamond. The phonon spectral energy density analysis reveals that MLP can effectively capture both the phonon mode softening and the linewidth broadening induced by the anharmonicity at finite temperatures in both materials. Compared to diamond, BAs exhibits a stronger anharmonicity revealed by the larger deviation from equilibrium position and more pronounced phonon broadening effect, especially at high temperatures. Furthermore, based on the phonon Boltzmann transport equation and three-phonon scattering process, our calculation results demonstrate that the accuracy of the MLP in predicting the κ is comparable to that of density-functional theory calculations for both diamond and BAs. However, this framework can only predict κ of diamond in agreement with experimental measurements, but significantly overestimates the κ of BAs compared to the experimental results, due to the significant impact of high-order phonon scattering process in BAs. In contrast, the κ values predicted by equilibrium molecular dynamics simulations combined with MLP agree well with experimental values for both BAs and diamond. Our study suggests that molecular dynamics simulation combined with MLP is a reliable and computationally efficient tool to account for full orders of anharmonicity and provide accurate predictions of material’s thermal conductivity without any a priori knowledge of the importance of high-order phonon anharmonicity.
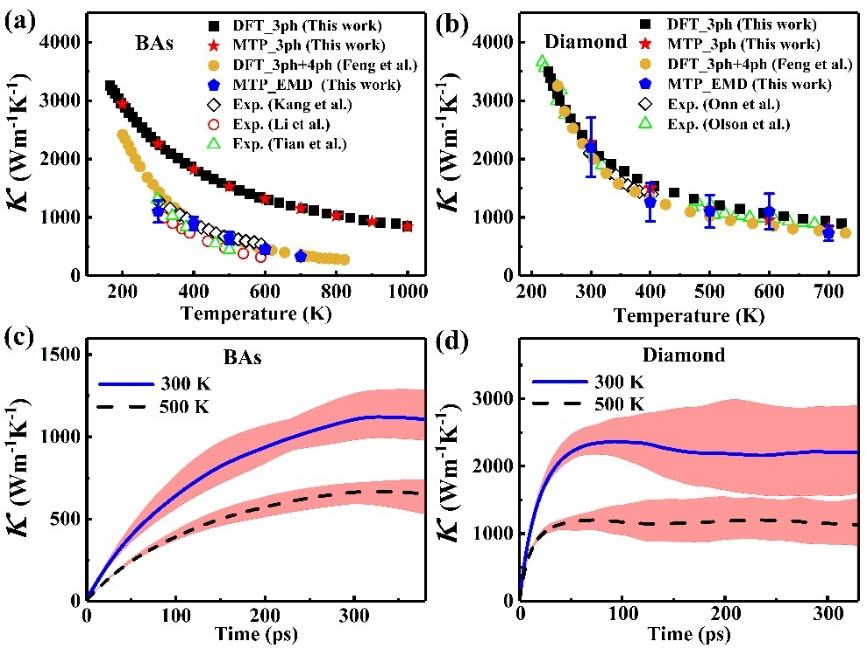
FIG. 1. Lattice thermal conductivity κ of BAs and diamond. (a) and (b) show, respectively, κ of BAs and diamond vs temperature from theoretical predictions (solid symbols) and experimental measurements (empty symbols). (c) and (d) show the predicted thermal conductivity by Green-Kubo method with MTP vs integration time for BAs and diamond, respectively. The solid and dashed lines represent the averaged thermal conductivity over 30 independent simulations, and the shadowed area represents the corresponding deviation.

